
�������: 1-15 ���鵽��ԭ�ӷ�������ѧ ��������ؼ�¼25�� . ��ѯʱ��(0.338 ��)
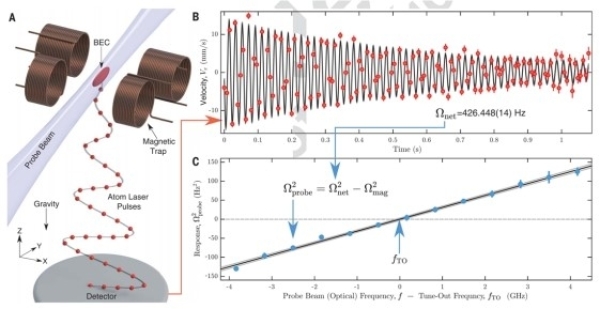
���գ��й���ѧԺ���ܲ�����ѧ�뼼�������о�Ժԭ�ӷ����ⳡ�������о�Ա�����ޡ����о�Ա�����۵ȣ���Ĵ����ǹ�����ѧ�����ô���ɯ��ѧ������ʵ���˺�ԭ��413 nm���㲨���ľ�ȷ�����;��ܲ����������ˡ����Ρ�ԭ�ӻ��㲨�����ܲ����������ӵ綯��ѧ��QED�����۵���;��������о��ɹ���Measurement of a helium tune-out frequency: an independent tes...

�й���ѧԺ������ѧ�����о����״�ʵ�ֳ�����ԭ�ӷ�Ӧ�ľ�ȷ����������ͼ��
�й���ѧԺ������ѧ�����о��� ������ԭ�ӷ�Ӧ ��ȷ�������
2020/9/28
���գ��������ӷ�Ӧ����ѧ�����ص�ʵ�����Ŷ���Ժʿ�Ŷ��ڳ�����ԭ�ӷ�Ӧ�Ķ���ѧ������ȡ���½�չ���״�ʵ�ֳ�����ԭ�ӷ�Ӧ�ľ�ȷ����������

�й���ѧ������ѧ�˽�ΰ��Է�������ڳ���ԭ������������ģ���о���ȡ����Ҫ��չ�������������������ʵ��ʵ��ԭ�������ȴ�»��ƵĻ����ϣ��ڹ⾧�����״�ʵ����1250��ԭ�Ӹ߱���Ⱦ���̬��ͬ���Ʊ���Ϊ���ڳ���ԭ�ӹ⾧��Ĺ�ģ������������ģ��춨�˻���������ʱ��6��19�գ���������ѧ���ڿ�����ѧ����־�ԡ�First Release����ʽ���߷����˸��о��ɹ���

��ǰ����ȫ������ԭ�����������רҵίԱ������ĵ����ȫ������ԭ�����������ѧ���������Ĵ�ʡ�˱��о��С�ɽ����ѧ̩ɽѧ������ȡ���֣�V���캣�ͻƺ�����ѧ���λᲢ���ᡰ�ر�����������
�����ܶȷ�������">=Gad#��������ԭ�Ӳ����������Чԭ��ʵ��"b1Hd#7TT ���飬��(��ԭ�Ӳ���8L5A00A<��ȫ���ӻ��飬�Ż���dLC) �ķ��ӽṹ���õ�����Ӧ��ƽ�⼸�ι���$ͬʱ�Ż���dL&I)C�ģ����ȶ��칹��$�Ƚ���������ˮ�������ڽ��룬�Ӷ���dL �γɸ��ȶ��Ľṹ������dL��Cԭ�ӵĵ縺�����ܴ��������ת��!���ӵ��ȶ�����ҪԴ��dL&C֮�������á����ݵ���A��������...

2010��12��19�գ����人��������ѧ�о����е����п�Ժ֪ʶ���¹�����Ҫ������Ŀ����ԭ�ӷ��������������������ջ����人�ٿ��������й���ѧԺ���ߵ�ԺУ�Լ�������Ȼ��ѧ����ί��ר�Һ��쵼�μӻ��顣���ר����������ȡ����Ŀ���ⱨ�漰�ĸ��ӿ��ⱨ���ȡ�õijɹ���ʾ�˳�ֵĿ϶�����������пϵ�����ͽ��顣����ĿΧ�Ƶ�ԭ�ӷ���������������ַ������人������������ԭ�ӡ����Ӻͺ˴Ź����о���������ƣ�ʵ����...
���ݺϷʹ�Դ���������Ҫ��Ϊ���кϷʹ�Դע����������λ�á�����Ⱥ���ɢ�ķ����ز�����������µ���������λ�ü������BPM���Ͱ˵缫������ɢ�������BESM��������������BPM�Ͱ˵缫BESMͨ�����ź���ȡx2��y2�Լ������ȵĹ�ʽ��ͨ�������缫��ʱ��Ƶ�ʷ������迹ƥ�������ȷ��������������ľ���������������CST microwave studio�а���������������...
�ԺϷʹ�Դ��ʹ�õ�ť��������λ�ü������������ز�����������������ˮƽ�ʹ�ֱ����������ȡ������ڷֱ��ʺʹ��书�ʽ����˹��㡣���Ż���ļ������װλ�ò���������λ�ý����˸߾�����ֵ���棬�Բ��������ò�ȺͺͶ����ȷ������д������õ�ֱ�۵�mappingͼ����϶���ʽ�����������źŽ��з�����ϣ�������λ�ƴӣ�0 mm��0 mm������4 mm��4 mm��ʱ����һ�����źŵı仯С��5��10��3��
ʹ��ͶӰ�������˦�����̽��������Դ��ͬ������µļ������ӣ�����ʵ���������˱Ƚϡ������������ͬ�������£�������̽��������Դ�ļ���������ʵ���������������һ�£��Ԧ����ǿ̶Ⱥ�ʵ�ʲ���ʱ����λ������IJ�ȷ���ȵ������вο���ֵ��
¬ɪ��ɢ�估��������ģ��
¬ɪ��ɢ�� ɢ����� �����ģ��
2011/8/22
��¬ɪ����ɢ����������г���ɢ�����ƫ�����������������о�������������ģ������������¬ɪ��ɢ�����ģ��������ģ���������������¬ɪ��ɢ�乫ʽ��С�Ƿ�ɢ�ɰ�ԭ�Ӽ以�����ڵ���������������ɣ��ں��С��û�������Ҳ������������ɢ�������£�ģ�����������¬ɪ��ɢ�乫ʽ�����Ľ��һ�¡�
������̬ģ���еĴ�ͳ����, ��̬���۵����Ľ��۵����̿��Կ�����һ��������������������̬����. �������һ���µ���̬���۵�����ȥ�۵������������·���, �ҽ��÷�������������̬���۵�����Y79W-W83F-Cu��ȥ�۵�������. ���ݷ�������, �ø÷�����õĵ���ȥ�۵������ܽ�����ǰ�����ķ�������ȷ.
���ܶȷ�������(DFT)��Becke 3LYP����,�ڲ�ͬ�����ϣ�6��31G��6��311Gϵ�У��¶�ƽ�нṹ��C 2h����T�νṹ(C2v)��CO2���������ab initio����.ͨ������,�õ���CO2������C2h��C2v���ֹ��͵Ľṹ�����������,��������CO2����������ȶ�����C2h��12����������ͼ.�������,CO2������������Ϊ2 kJ•mol��1,CO2����֮����Ƶ...
����Gaussian98����HF��MP2��G2�����ͳ�cc-pVQZ�����Դ������ӽ����˼����Ż��͵��������������������ʵ�����ݽ����˱Ƚϡ�
����ȫ������Զ���̬Dirac-Fockƽ���ܼ�����ϵͳ�������˸߰�����������3s2S��3d2D(Z=14��103�����ļ���E2����ԾǨ���ܼ������ԾǨ���ʺ�����ǿ�ȣ������п����˺˵��������ЧӦ��Breit������QED���������ý���������ʵ�����ݼ���������ֵ�����˱Ƚϣ����������������ԭ�������ĸߺɵ����ӵĵ��ļ���E2����ԾǨ��ԾǨ���ʺ�����ԭ�ӵĵ�ż��E1���൱����ICF��MCF���¸��ܶȼ�...
���������Ť�����������Ƽӽ�������������̬ƽ�������£�ϵͳ�����������⡢�����������n��6�ĸ���̬֮�����ײ�������̽��棬�������е����۽����������ϸ�ĶԱȷ�������������������Ť�������Ƶ�����������ϵúܺã����ƫ��һ�㶼С��10%������û�п��ǹ���ЧӦ�������Ľ����ǿ��Ϸ��������������������������ϵ͵�������нϴ�ƫ������������ϽϺã����ƫ��һ����15%���ڡ��÷������Է��������...
�й��о����������а�-��
- ���ڼ���...
�й�ѧ���ڿ����а�-��
- ���ڼ���...
�����ѧ���л������а�-��
- ���ڼ���...
�й���ѧ���а�-��
- ���ڼ���...
�ˡ���-ƪ
- ���ڼ���...
�Ρ���-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
�������� -ƪ
- ���ڼ���...
֪ʶҪ��-ƪ
- ���ڼ���...
���ʶ�̬-ƪ
- ���ڼ���...
��������-ƪ
- ���ڼ���...
ѧ��ָ��-ƪ
- ���ڼ���...
ѧ��վ��-ƪ
- ���ڼ���...