
搜索结果: 1-7 共查到“物理学 第一性原理计算”相关记录7条 . 查询时间(0.443 秒)
2019年4月8日-13日,第二届“手征有效场论与原子核第一性原理计算国际学术会议”在南京市钟山宾馆召开。本次会议由南京航空航天大学、四川大学和北京大学共同主办,我校材料科学与技术学院核科学与技术系承办;会议得到了国家自然科学基金委和中国高等科学技术中心的支持,旨在为海内外该领域的物理学家搭建交流平台,推动我国在手征有效场论与原子核第一性原理计算这些方面的研究。4月9日上午,会议在南京市钟山宾馆开...
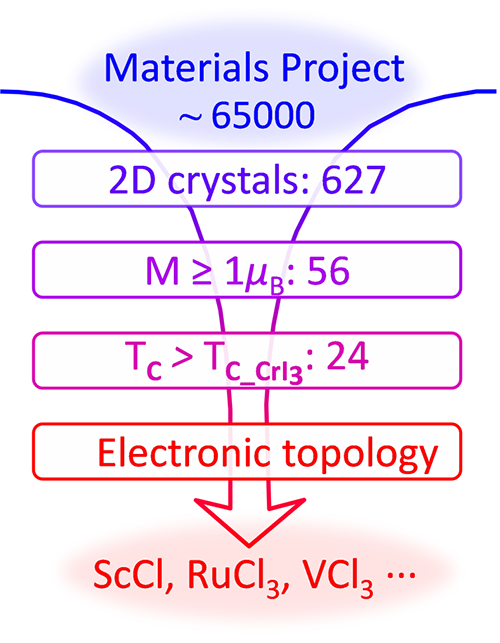
中国科学院物理研究所第一性原理计算筛选本征二维磁性材料研究取得进展(图)
中国科学院物理研究所 第一性 原理计算 二维磁性材料研
2018/12/20
近年来,基于第一性原理的高通量筛选方法逐渐成为发现新材料的途径,为寻找本征二维磁性材料及研究新奇电子性质提供了新手段。最近,中国科学院物理研究所/北京凝聚态物理国家研究中心表面物理国家重点实验室SF10组博士研究生刘行在副研究员孙家涛和研究员孟胜的指导下,搭建了高通量筛选二维磁性材料的第一性原理计算流程及相关数据库,从材料数据库中发现了数十种新型层状磁性材料。一些具有高居里温度的铁磁材料还呈现出新...
Li3Bi结构、力学和电子性质的第一性原理计算
Li3Bi 各向异性 第一性原理
2012/11/5
基于密度泛函的线性响应理论,通过第一性原理的赝势方法,对Fm-3m相Li3Bi的结构、力学和电子性质做了系统的研究.优化得到的平衡结构参数与实验值符合得很好.计算表明,Fm-3m结构在零压下的焓最低,满足力学稳定标准,是最稳定的结构.计算得到的Fm-3m相Li3Bi的块体模量、剪切模量和弹性模量分别为30.2,25.5 GPa和59.6 GPa.德拜温度是312 K.Li3Bi具有小的弹性各向异性...
常压下Se和Te电子结构和弹性性质的第一性原理计算
第一性原理计算 能带结构 弹性系数 德拜温度
2012/11/14
利用基于密度泛函的第一性原理, 计算ⅥA族元素Se和Te在常压下的能带结构、 电子态密度、 弹性系数和德拜温度. 能带结构和电子态密度的计算结果表明: Se为间接带隙半导体, Se费米面附近的导带和价带主要来自外层4p4电子的贡献, 4s2电子对费米面附近的导带和价带贡献较少; Te为直接带隙半导体, Te费米面附近的导带和价带主要来自外层5p4电子的贡献, 5s2电子对费米面附近的导带和价带贡...
通过建立的匹配生长超晶胞模型,研究了Al2O3/SiO2纳米异质薄膜的结构特点和电子结构.通过第一性原理计算,并与单一相的Al2O3薄膜表面进行了对比,讨论了相关的结构变化.通过分析原子位置、键长和键角的改变,研究了这种Al2O3单层膜的电子结构和化学成键.
In2O3电子结构与光学性质的第一性原理计算
In2O3 光学性质 电子结构 第一性原理
2009/10/13
采用基于密度泛函理论框架下的第一性原理平面波超软赝势方法, 计算了In2O3电子结构和光学线性响应函数, 系统研究了In2O3电子结构与光学性质的内在关系. 利用计算的能带结构和态密度分析了带间跃迁占主导地位的In2O3材料的能量损失函数、介电函数、反射图谱, 根据电荷密度差分图分析了In2O3材料的化学和电学特性. 研究结果表明In2O3光学透过率在可见光范围内高达85%, 可作为优异的透明导电...
Mn掺杂ZnO光学特性的第一性原理计算
密度泛函理论(DFT) 第一性原理 超软赝势 Mn掺杂ZnO
2008/5/19
采用基于密度泛函理论(DFT)的第一性原理平面波超软赝势方法计算了纤锌矿ZnO及不同量Mn 掺杂ZnO 晶体的电子结构,分析了掺杂对ZnO 晶体的能带结构、电子态密度、差分电荷分布的影响. 计算结果表明,随着Mn 掺杂含量的增加,ZnO 禁带宽度相应增加并且对紫外吸收区的光吸收能力也随之增强.